{kind=link}
AI has proven to be a force multiplier, helping to create a future where scientists can design entirely new materials, while engineers seamlessly transform these designs into production plans—all without ever setting foot in a lab. As AI continues to redefine the boundaries of innovation, this once elusive vision is now more within reach.
Recognizing this paradigm shift, a convergence of expertise is emerging at the intersection of AI, chemistry, and material science. This interdisciplinary collaboration between AI experts, chemists, and material scientists aims to transcend the traditional, difficult-to-scale trial-and-error methods.
Together, they are building AI models and datasets to shorten the design-to-production cycle from a decade to months, with the aspiration to enable next-generation innovations from efficient batteries to biodegradable polymers.
This post introduces the NVIDIA ALCHEMI (AI Lab for Chemistry and Materials Innovation), which aims to accelerate chemical and material discovery with AI.
AI-accelerated chemical and materials discovery workflow
The discovery of a novel material can be broadly divided into four stages:
- Hypothesis generation
- Solution space definition
- Property prediction
- Experimental validation
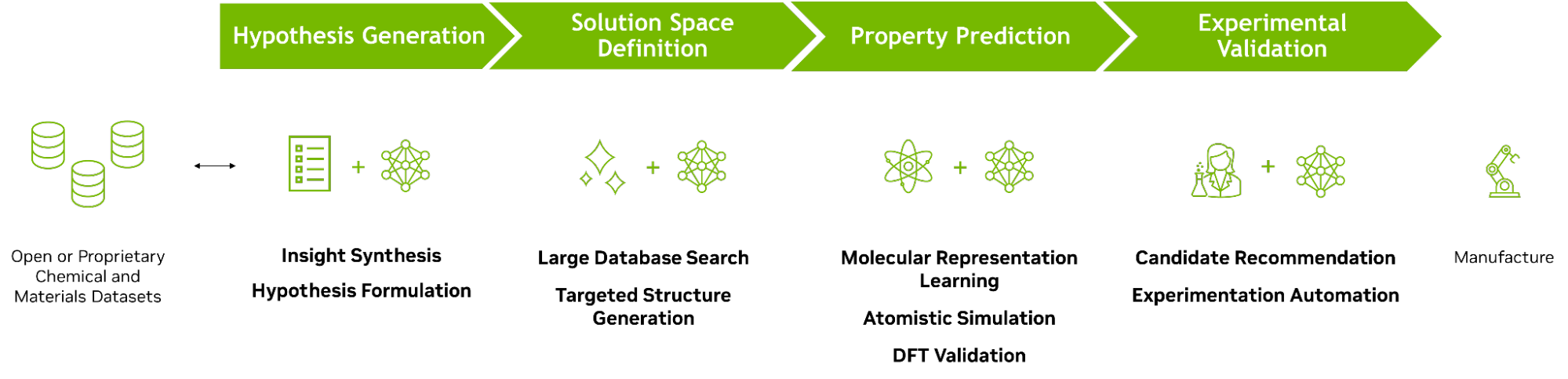
Hypothesis generation
- Insight synthesis: Prompt chemistry-informed large language models (LLMs) (that is, LLMs trained or fine-tuned with chemistry literature) to synthesize vast corpus of chemical literature.
- Hypothesis formulation: Leverage chemistry-informed LLMs as thought partners to formulate and refine hypotheses, drawing on the LLM ability to draw connections between seemingly unrelated concepts.
Solution space definition
- Large database search: Identify subspace of interest from existing chemical databases.
- Targeted structure generation: Employ generative AI to propose new-to-science candidates based on desired properties.
Property prediction
- Molecular representation learning: Leverage learned molecular representations to predict properties of candidates in solution space.
- Atomistic simulation: Apply AI surrogates (MLIPs, GNNs) to predict high-fidelity properties.
- Density Functional Theory (DFT) Validation: Validate predicted properties through DFT simulations.
Experimental validation
- Candidate recommendation: Propose candidates to validate in the lab (for example, through Bayesian optimization to balance between exploitation of known chemistry versus exploration of white space).
- Experimentation automation: Expedite lab synthesis and testing with AI-enabled self-driving lab and active learning.
Accelerating R&D in chemistry and materials science
NVIDIA ALCHEMI is dedicated to accelerating R&D in chemistry and materials science through the power of AI. ALCHEMI will include APIs and NVIDIA NIM-accelerated inference microservices for developers and researchers, enabling:
- Creation and deployment of generative AI models to explore the vast materials universe and suggest new potential candidates based on desired properties.
- Development and utilization of AI surrogate models that strike a balance between accuracy and computational cost compared to traditional ab initio simulation methods like Density functional theory (DFT).
- Accessible chemical and materials Informatics tooling and pre-trained foundation models for mapping materials representations to properties for fast screening.
- Simulation tools for generating synthetic datasets to train and fine-tune AI models for new use cases.
NVIDIA ALCHEMI is introducing its first accelerated materials discovery NIM, focused on accelerating a common but important application of a class of AI surrogate model called machine learning interatomic potentials (MLIPs).
Machine learning interatomic potentials
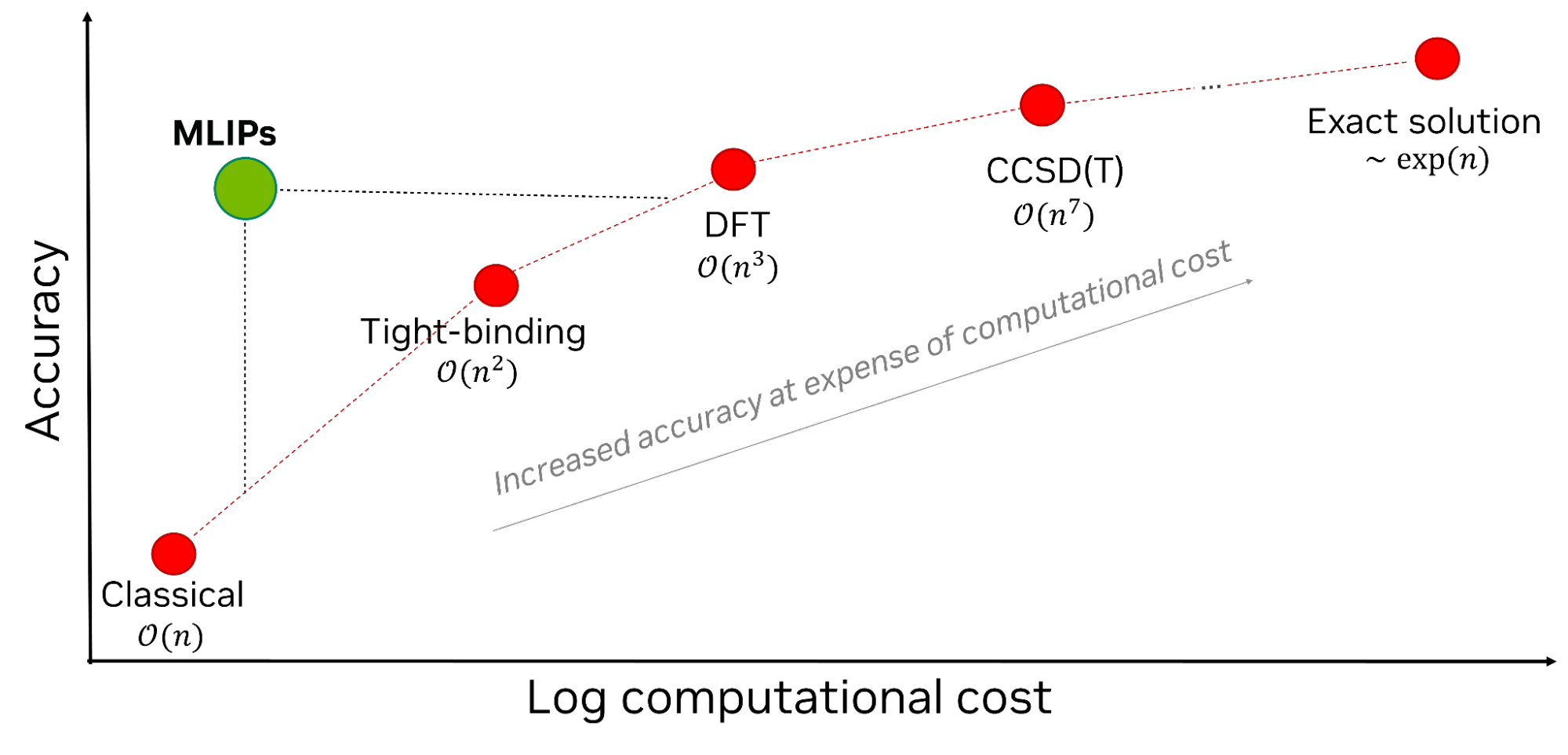
Before diving into the NIM, it is helpful to first introduce a few background concepts. A range of traditional computational methods exist, from classical physics to exact solutions. Each method comes with a trade-off between accuracy and computational cost. A method with higher accuracy, such as DFT, comes with significantly higher computational cost. By way of illustration, DFT calculation for a system with 10 atoms takes minutes, while that of 100 atoms takes hours. However, for a system with 1,000+ atoms, DFT (which scales cubically with number of atoms) can take weeks, rendering it impractical for large complex systems (nanoparticles with 102 to 106 atoms, proteins with 104 to 105 atoms, for example) or as a means to high-throughput simulation.
MLIPs offer the best for both accuracy and computational cost by leveraging AI to map atomic structures to potential energies and atomic forces (Figure 2). MLIPs often use GNNs to represent atomic structures as graphs, with atoms represented as nodes and interatomic distances within a preset cutoff radius as edges. This enables the GNNs to readily capture spatial relationships between atoms that make up a molecule or material.
MLIPs have widespread applications in chemistry, material science, and biology, from material property predictions to large-scale molecular dynamics simulations. This contrasts with physics-informed neural networks (PINNs) that embed knowledge from physics equations and have relevance primarily in computational fluid dynamics.
Geometry relaxation
In many chemical and material discovery workflows, a key task is to differentiate stable materials from unstable ones through geometry relaxation and energy comparison. This is especially crucial in workflows involving generative models, as the generated candidates may not be inherently stable.
During geometry relaxation, a material’s energy is minimized by iteratively evaluating the forces on each atom (inference) and adjusting atomic positions in the direction of the bottom of potential energy well (optimization). Each material candidate may require thousands of relaxation steps to achieve this energy minimum. Stable materials, identified by comparing their energies to other candidates, are then prioritized for further evaluation based on their desired properties.
NVIDIA Batched Geometry Relaxation NIM
While MLIPs accelerate energy and force calculations for geometry relaxation significantly (versus full DFT), the actual implementation can still be time-consuming. This is why NVIDIA has developed the NVIDIA Batched Geometry Relaxation NIM to accelerate geometry relaxation calculations. An NVIDIA NIM is simply a container that exposes an API. This NIM focuses on accelerating geometry relaxation of potential battery materials, using MACE-MP-0 and AIMNet2 models respectively in Atomic Simulation Environment (ASE). Traditional CPU-based simulation workloads only partially utilize the full performance of MLIPs and incur significant communication penalties in CPU-GPU data movement. Moreover, MLIPs typically underutilize the GPU, as they only process a single system at a time.
By leveraging NVIDIA Warp, a Python developer framework for writing GPU-accelerated simulation code, you can write regular Python functions and have Warp compile them at runtime into efficient GPU kernel code. Rather than doing one geometry relaxation at a time, you can launch batches of geometry relaxation simulations, enabling hundreds of energy minimizations to run in parallel efficiently and maximizing the usage of available GPU resources. This is critical, since each candidate requires thousands of relaxation steps.
Table 1 shows results for 2,048 small to medium inorganic crystal systems (20-40 atoms per periodic cell), using the MACE-MP-0 model and ASE FIRE geometry optimizer on a single NVIDIA H100 80 GB GPU. The same 2,048 samples without the NVIDIA Batched Geometry Relaxation NIM take ~15 minutes versus 36 seconds with the NIM, a ~25x acceleration. Increasing the batch size from 1 to 128 accelerates the geometry relaxation further to 9 seconds, representing a ~100x acceleration.
| Batched Geometry Relaxation NIM | Batch size | Total time (s) | Average time per system (s/system) | Approximate speedup |
| Off | 1 | 874 | 0.427 | 1x |
| On | 1 | 36 | 0.018 | 25x |
| On | 128 | 9 | 0.004 | 100x |
Table 2 shows comparable acceleration for 851 small to medium organic molecules (~20 atoms per molecule) from the GDB-17 database, using AIMNet2 and ASE FIRE. Turning on the NIM results in a ~60x acceleration from ~11 minutes to 12 seconds. Further acceleration is observed from increasing batch size 1 to 64, representing a ~800x acceleration. Differences in observed speed-up (100x with MACE-MP-0 versus 800x with AIMNet2) arise from the modeled systems (periodic crystals versus small molecules), rather than the models deployed.
| Batched Geometry Relaxation NIM | Batch size | Total time (s) | Average time per system (s/system) | Approximate speedup |
| Off | 1 | 678 | 0.796 | 1x |
| On | 1 | 12 | 0.014 | 60x |
| On | 64 | 0.9 | 0.001 | 800x |
SES AI, a leading developer of lithium-metal batteries, is exploring using the NVIDIA ALCHEMI NIM microservice with the AIMNet2 model to accelerate the identification of electrolyte materials used for electric vehicles.
“SES AI is dedicated to advancing lithium battery technology through AI-accelerated material discovery, using our Molecular Universe Project to explore and identify promising candidates for lithium metal electrolyte discovery,” said Qichao Hu, CEO of SES AI. “Using the ALCHEMI NIM microservice with AIMNet2 could drastically improve our ability to map molecular properties, reducing time and costs significantly and accelerating innovation.”
SES AI recently mapped 100,000 molecules in half a day, with the potential to achieve this in under an hour using ALCHEMI, signaling how the microservice is poised to have a transformative impact on material screening efficiency.
Looking ahead, SES AI aims to map the properties of up to 10 billion molecules within the next couple of years, pushing the boundaries of AI-driven high-throughput discovery.
Get started with the NVIDIA Batched Geometry Relaxation NIM
To get started with the Batched Geometry Relaxation NIM, you’ll need the following:
- Familiarity with Python and ASE
- Knowledge of running a Docker container
- Need for MACE-MP-0 (materials) or AIMNet2 (molecules) model
Procedure
It’s important to understand that an NVIDIA NIM is simply a container that exposes an API. Interacting with the NIM involves two key steps:
First, launch the NIM container:
docker run --rm -it --gpus all
-p 8003:8003 \
<NIM container address>
Note that --gpus all directs which devices the NIM will load separate instances of the model and optimizer on. If more than one device exists, the NIM will distribute requests among the available model instances. Second, we expose the port forwarding 8003:8003, mapping local port 8003 to container port 8003, which is the default communication port of the NIM API. The requests from the client get routed to this port.
Next, with the container launched and instantiated, you can now submit requests:
import os, requests, json
import numpy as np
from ase import Atoms
from ase.io.jsonio import MyEncoder, decode
atoms: list[Atoms] = # This is your ase.Atoms input molecules
# Define the url of the NIM
# below is a typical local IP address and port
url: str = 'http://localhost:8003/v1/infer'
# Prepare input atoms by converting to json
data = {"atoms": json.dumps(atoms, cls = MyEncoder)}
headers = {'Content-Type': "application/json"}
# Submit request to NIM
response = requests.post(url, headers = headers, json=data)
response.raise_for_status()
# Convert from json output back to ase.Atoms
optimized_atoms = decode(response.json())
Notes to consider:
- Given that ASE is a popular molecular modeling python package, the input data is expressed in the form of
ase.Atoms, which is a flexible way to describe material systems. - The user must provide the URL of the NIM. This URL, http://localhost:8003/v1/infer, comprises three components:
- IP address:
localhostin this example - Port:
8003was mapped in the Docker command - NIM inference API endpoint:
v1/infer
- IP address:
- The user converts the
ase.Atomsto a .json string usingaseutilities and submits a request using therequestslibrary. This post request transmits the data to the Batch Geometry Relaxation NIM and starts the relaxation process, asynchronous from the client code. When the molecules or materials have converged to a relaxed state, they are transmitted back to the client and can be converted back toase.Atomsas shown in the code snippet above.
Conclusion
Using the NVIDIA Batched Geometry Relaxation NIM resulted in a 800x acceleration in MLIP calculations. This acceleration, close to three orders of magnitude, opens the door to high-throughput simulations of millions of candidates, enabling next-generation foundation models trained with high-quality data and improving downstream property prediction capabilities. It also enables simulation of more complex and realistic systems, unlocking new chemistries and applications. This is just the beginning. Through ALCHEMI, NVIDIA aims to accelerate AI-enabled chemical and material discovery workflow end-to-end, and usher in a new era of breakthrough discoveries powering a more sustainable, healthier future.
Sign up to receive notification when the NVIDIA Batched Geometry Relaxation NIM is available for download.